
本文系统阐述了如何利用 charmm-gui.org 生成 Gromacs 输入文件,并通过精细化模拟方案完成分子动力学模拟的全过程。初始结构文件源自 Schrödinger Maestro 的 Glide Dock 模块,包含蛋白质-配体复合物结构。鉴于小分子力场参数生成的准确性对模拟结果具有决定性影响,本文特别强调了该环节的技术要点。
一、初始结构预处理
1.1 小分子结构提取
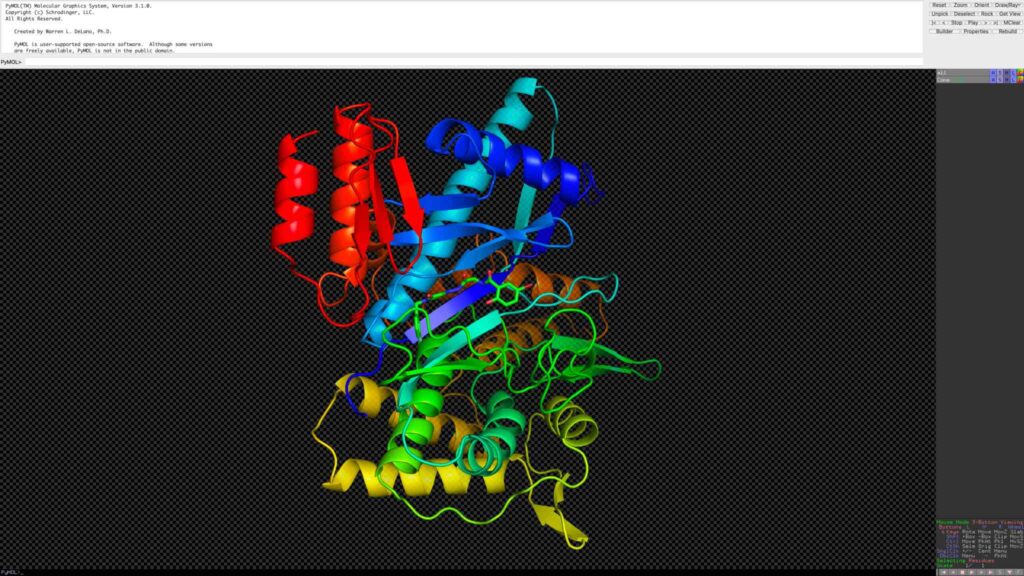
在 PyMOL 中打开复合物结构,选取小分子并另存为 LIG.sdf 文件。
1.2 力场参数生成
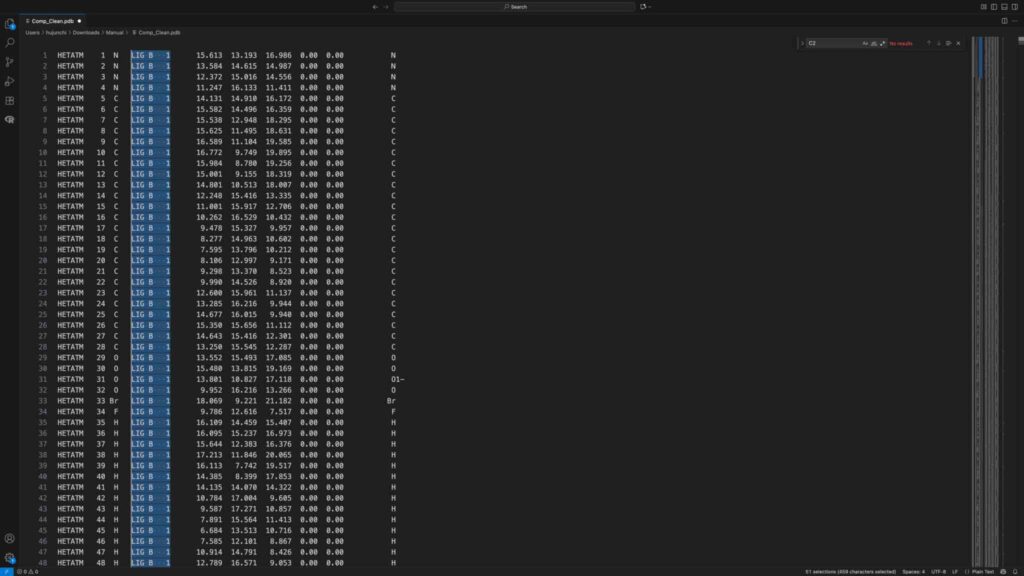
在 CHARMM-GUI 中的 Input Generator 的 Ligand Reader & Modeler 模块中导入 LIG.sdf 文件,逐一核对分子中原子与键的连接情况,特别注意氢原子数量。检查完成后,点击保存下载配体 .sdf 文件。
1.3 结构叠合与复合物准备
使用 PyMOL 导入刚才下载的配体 .sdf 文件,逐一选择配体中的原子,并将选择图层重命名为 sele1。然后选中复合物对应的原子位置,并使用以下语句将配体叠合至正确位置:
center2 = cmd.get_model("sele").atom[0].coord;cmd.alter_state(0, "sele1", f"x={center2[0]}; y={center2[1]}; z={center2[2]}")将配体叠合至小分子结合位点,选中受体文件和小分子,另存为 Comp_Clean.pdb。检查 Comp_Clean.pdb 文件,修改小分子配体名称、链和 ID:
二、CHARMM-GUI参数生成
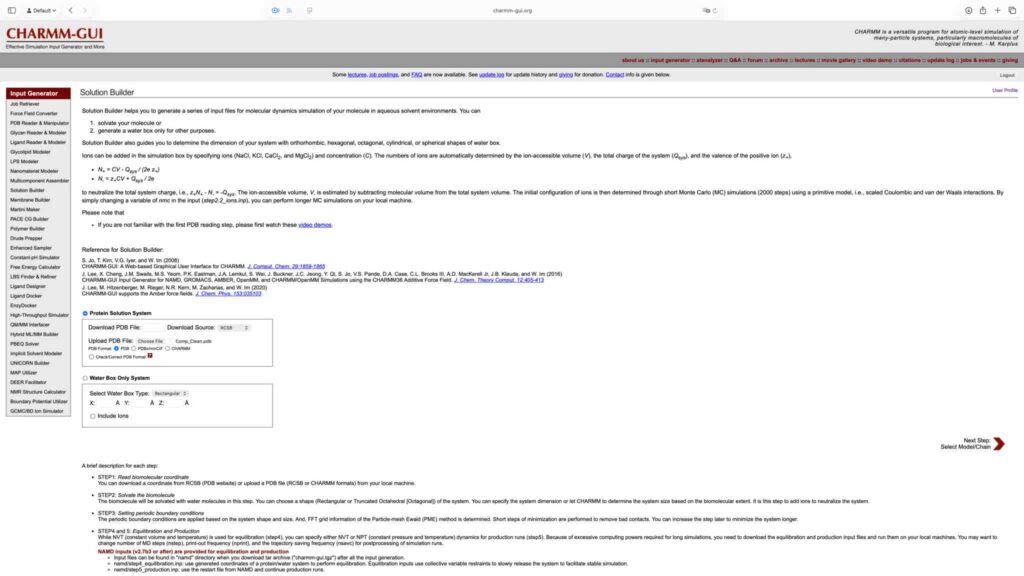
2.1 Solution Builder 模块配置
鉴于这里所用复合物为溶液环境下的蛋白质,因此参数生成采用 Solution Builder 模块。上传复合物 .pdb 文件,并进入下一步:
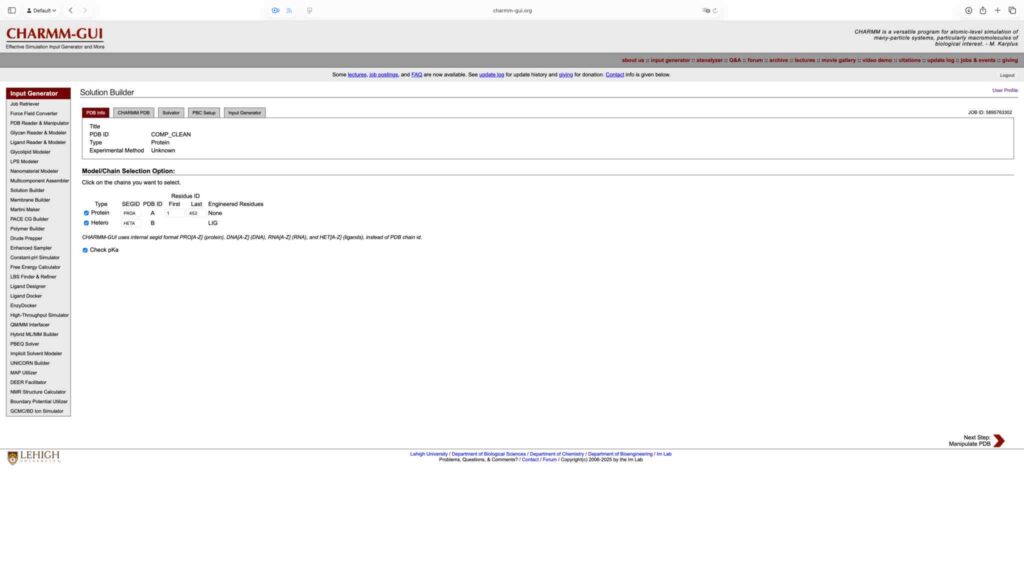
选中蛋白和 LIG,勾选 pKa 检查,然后点击 Next Step。
2.2 力场文件生成
检测到小分子需要力场文件,因此使用 CHARMM 根据刚才下载的 .sdf 文件生成力场。保持其他参数默认,点击 Next 继续。
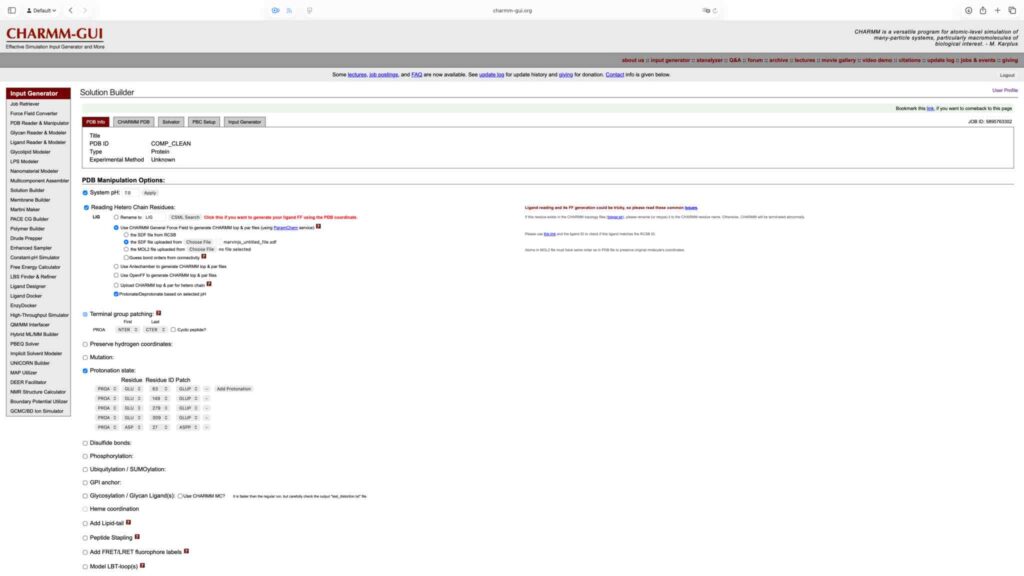
2.3 动力学参数文件生成
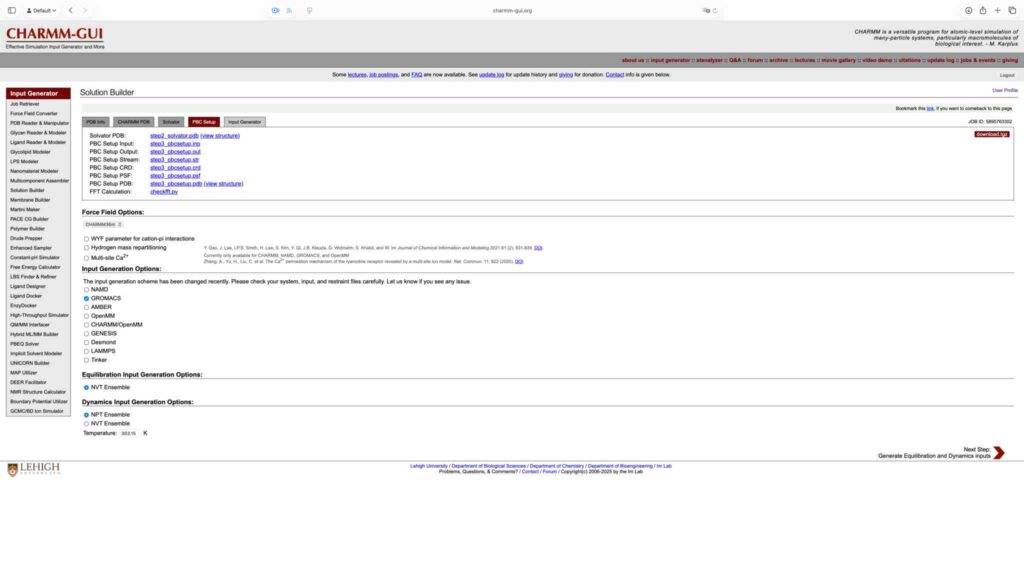
将溶剂盒形状设置为正八面体,水盒与溶质边缘保持 10 Å 距离。将离子浓度调整为 0.15 M,重新计算所需离子数量后进入下一步。
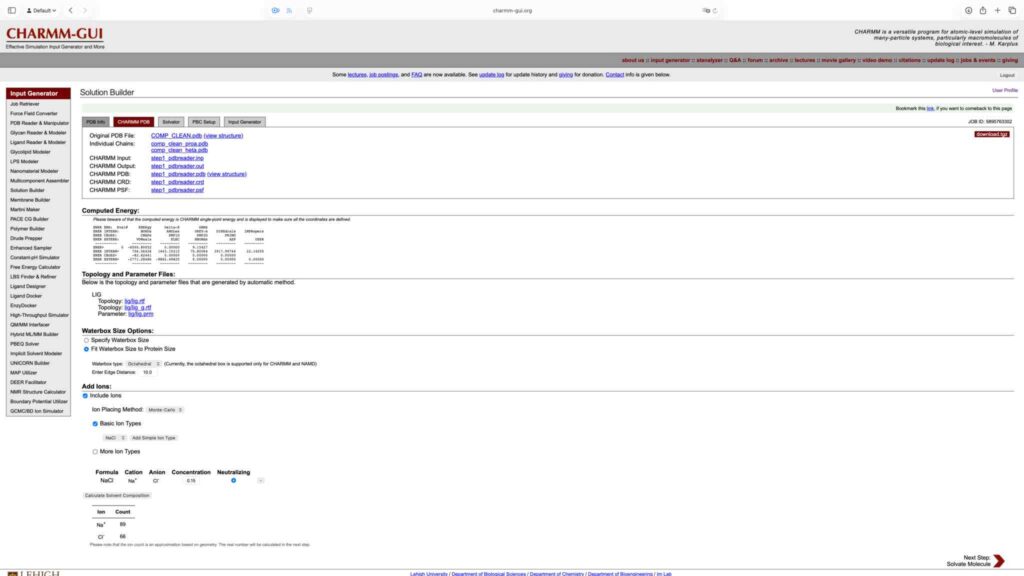
保持默认值,继续下一步:
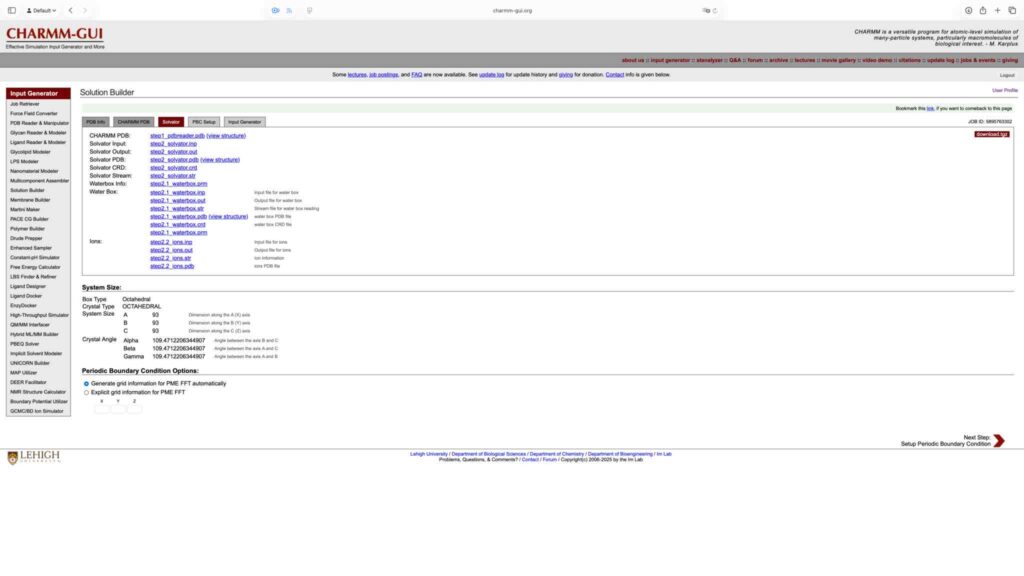
选择生成 Gromacs 格式的输入文件,保持其他参数默认,点击 Next Step 继续。
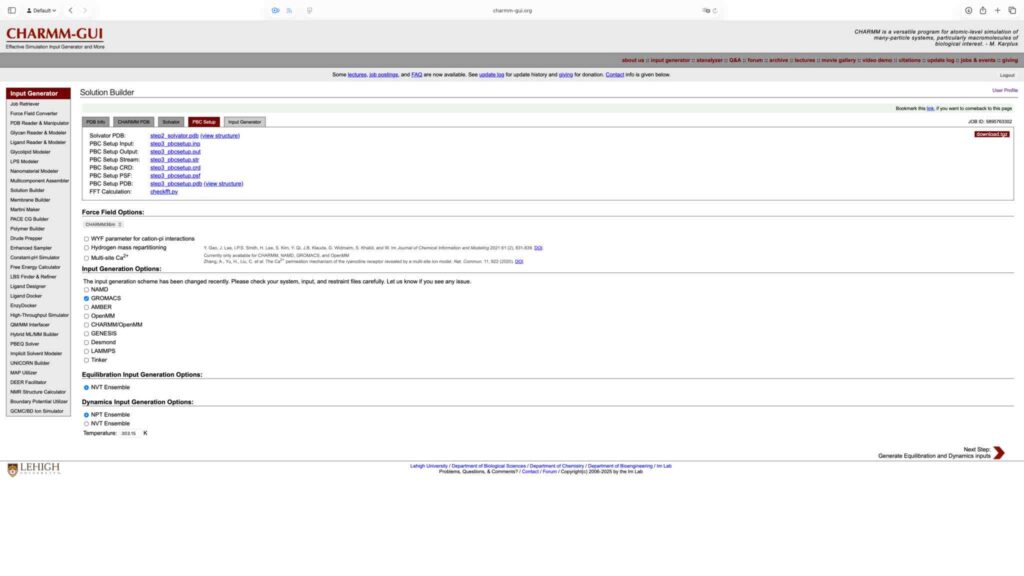
点击 download.tgz 下载生成文件,完成基于 Charmm-GUI 的 Gromacs 运行文件准备工作:
三、Gromacs 输入文件准备
3.1 文件解压与目录结构
解压并进入下载的 .tar 文件:
tar -zxvf charmm-gui.tar && cd charmm-gui-*可以看到当前文件夹下有一个 gromacs 文件夹,所有的 gromacs 运行所需的文件均在此文件夹下:
(base) hujunchi@charmm-gui-5895763302 % cd gromacs
(base) hujunchi@gromacs % ls
index.ndx step3_input.gro step3_input.psf step4.1_equilibration.mdp topol.top
README step3_input.pdb step4.0_minimization.mdp step5_production.mdp toppar3.2 模拟阶段划分
CHARMM-GUI 为我们提供了三个运行步骤的文件:能量最小化文件(step4.0_minimization.mdp)、短期平衡文件(step4.1_equilibration.mdp)和动力学生产文件(step5_production.mdp),其中 step4 为体系准备阶段,step5 为分子动力学生产阶段。然而,CHARMM-GUI 默认的准备方案相对粗略,容易在初期引入较大应力或结构破坏,尤其对于大体系或含配体体系更为明显。为此,我们设计了更精细的能量最小化与逐步放松方案,其核心思路为固定大部分结构并逐步解除约束,流程如下:
能量最小化(溶剂 → 全体系)→ 短期恒容平衡(溶剂 → 蛋白氢原子 → 蛋白侧链 → 全蛋白 → 全体系)→ 短期恒压平衡(溶剂 → 蛋白氢原子 → 蛋白侧链 → 全蛋白 → 全体系)。
四、动力学过程实施
4.1 能量最小化阶段
4.1.1 原子约束文件生成
为了设置原子限制条件,需要先生成蛋白和配体的 .gro 文件:
gmx_mpi trjconv -f step3_input.gro -s step3_input.gro -o PROA.gro #选择 1(Protein)
gmx_mpi trjconv -f step3_input.gro -s step3_input.gro -o LIG.gro #选择 13(LIG)使用 genrestr 命令:
gmx_mpi genrestr -f PROA.gro -o toppar/PROA_S.itp -fc 1000 1000 1000 #选择 0(System)
gmx_mpi genrestr -f PROA.gro -o toppar/PROA_dH.itp -fc 1000 1000 1000 #选择 2(Protein-H)
gmx_mpi genrestr -f PROA.gro -o toppar/PROA_M.itp -fc 1000 1000 1000 #选择 5(MainChain)
gmx_mpi genrestr -f LIG.gro -o toppar/LIG_S.itp -fc 1000 1000 1000 #选择 0(System)4.1.2 拓扑文件修改
修改 topol.top 文件,在力场部分增加限制性条件:
; Include forcefield parameters
#include "toppar/forcefield.itp"
#include "toppar/PROA.itp"
#ifdef POSRE_PROA
#include "toppar/PROA_S.itp"
#endif
#ifdef POSRE_PROH
#include "toppar/PROA_dH.itp"
#endif
#ifdef POSRE_PROM
#include "toppar/PROA_M.itp"
#endif
#include "toppar/LIG.itp"
#ifdef POSRE_LIG
#include "toppar/LIG_S.itp"
#endif
#include "toppar/SOD.itp"
#include "toppar/CLA.itp"
#include "toppar/TIP3.itp"4.1.3 分阶段能量最小化
删除 step4.0_minimization.mdp 并新建 step4.0.1_minimization_solvent.mdp:
rm step4.0_minimization.mdp
touch step4.0.1_minimization_solvent.mdp修改 step4.0.1_minimization_solvent.mdp 内容如下:
integrator = steep ; 最速下降法
emtol = 1000.0 ; 收敛力阈值 (kJ/mol/nm)
emstep = 0.01 ; 最大步长 (nm)
nsteps = 5000 ; 最大步数
; Neighbor searching
nstlist = 20
cutoff-scheme = Verlet
rlist = 1.2
; Electrostatics
coulombtype = PME
rcoulomb = 1.2
; van der Waals
vdwtype = Cut-off
rvdw = 1.2
vdw-modifier = Force-switch
rvdw-switch = 1.0
; Constraints
constraints = h-bonds
constraint_algorithm = LINCS
; Position restraints
define = -DPOSRE_PROA -DPOSRE_LIG ; 使用限制性 .itp 文件固定蛋白和配体生成 step4.0.1_minimization_solvent 的预处理文件:
gmx_mpi grompp -c step3_input.gro -r step3_input.gro -f step4.0.1_minimization_solvent.mdp -p topol.top -o step4.0.1_minimization_solvent.tpr运行 step4.0.1_minimization_solvent 步骤,并等待运行完成:
mpirun -np 16 gmx_mpi mdrun -deffnm step4.0.1_minimization_solvent同理,创建 step4.0.2_minimization_all.mdp,内容如下:
integrator = steep
emtol = 1000.0
emstep = 0.01
nsteps = 5000
; Neighbor searching
nstlist = 20
cutoff-scheme = Verlet
rlist = 1.2
; Electrostatics
coulombtype = PME
rcoulomb = 1.2
; van der Waals
vdwtype = Cut-off
rvdw = 1.2
vdw-modifier = Force-switch
rvdw-switch = 1.0
; Constraints
constraints = h-bonds
constraint_algorithm = LINCS
; Position restraints
; 不使用位置约束,完全放松体系生成 step4.0.2_minimization_all 的预处理文件:
gmx_mpi grompp -c step4.0.1_minimization_solvent.gro -r step4.0.1_minimization_solvent.gro -f step4.0.2_minimization_all.mdp -p topol.top -o step4.0.2_minimization_all.tpr运行 step4.0.2_minimization_all 步骤,并等待运行完成:
mpirun -np 16 gmx_mpi mdrun -deffnm step4.0.2_minimization_all4.2 平衡阶段
4.2.1 恒容平衡阶段
完成能量最小化后,就可以进行短期恒容平衡了,第一步先平衡溶剂分子,新建 step4.1.1_solvent_relaxation.mdp:
integrator = md
nsteps = 100000 ; 100 ps
dt = 0.001
; Output control
nstxout-compressed = 5000
nstxout = 0
nstvout = 0
nstfout = 0
nstcalcenergy = 100
nstenergy = 500
nstlog = 500
; Neighbor searching
cutoff-scheme = Verlet
nstlist = 20
rlist = 1.2
; Electrostatics / VDW
coulombtype = PME
rcoulomb = 1.2
vdwtype = Cut-off
vdw-modifier = Force-switch
rvdw-switch = 1.0
rvdw = 1.2
; Temperature coupling (moderate)
tcoupl = V-rescale
tc-grps = SOLU SOLV
tau_t = 1.0 1.0
ref_t = 303.15 303.15
; Parallel/comm
nstcomm = 100
comm_mode = linear
comm_grps = SOLU SOLV
; Velocities (only if starting new NVT)
gen-vel = yes
gen-temp = 303.15
gen-seed = -1
; Constraints
constraints = h-bonds
constraint_algorithm = LINCS
; Pressure coupling
pcoupl = no
; Position restraints
define = -DPOSRE_PROA -DPOSRE_LIG生成并运行 step4.1.1_solvent_relaxation:
gmx_mpi grompp -c step4.0.2_minimization_all.gro -r step4.0.2_minimization_all.gro -n index.ndx -f step4.1.1_solvent_relaxation.mdp -p topol.top -o step4.1.1_solvent_relaxation.tpr
nohup mpirun -np 16 gmx_mpi mdrun -deffnm step4.1.1_solvent_relaxation &新建 step4.1.2_hydrogen_relaxation.mdp:
integrator = md
nsteps = 100000 ; 100 ps
dt = 0.001
; Output control
nstxout-compressed = 5000
nstxout = 0
nstvout = 0
nstfout = 0
nstcalcenergy = 100
nstenergy = 500
nstlog = 500
; Neighbor searching
cutoff-scheme = Verlet
nstlist = 20
rlist = 1.2
; Electrostatics / VDW
coulombtype = PME
rcoulomb = 1.2
vdwtype = Cut-off
vdw-modifier = Force-switch
rvdw-switch = 1.0
rvdw = 1.2
; Temperature coupling
tcoupl = V-rescale
tc-grps = SOLU SOLV
tau_t = 0.5 0.5
ref_t = 303.15 303.15
; Parallel/comm
nstcomm = 100
comm_mode = linear
comm_grps = SOLU SOLV
; Velocities
gen-vel = no
; Constraints
constraints = h-bonds
constraint_algorithm = LINCS
lincs_iter = 1
lincs_order = 4
; Pressure coupling
pcoupl = no
; Position restraints
define = -DPOSRE_PROH -DPOSRE_LIG生成并运行 step4.1.2_hydrogen_relaxation:
gmx_mpi grompp -c step4.1.1_solvent_relaxation.gro -r step4.1.1_solvent_relaxation.gro -n index.ndx -f step4.1.2_hydrogen_relaxation.mdp -p topol.top -o step4.1.2_hydrogen_relaxation.tpr
nohup mpirun -np 16 gmx_mpi mdrun -deffnm step4.1.2_hydrogen_relaxation &新建 step4.1.3_sidechain_relaxation.mdp:
integrator = md
nsteps = 100000 ; 100 ps
dt = 0.001
; Output control
nstxout-compressed = 5000
nstxout = 0
nstvout = 0
nstfout = 0
nstcalcenergy = 100
nstenergy = 500
nstlog = 500
; Neighbor searching
cutoff-scheme = Verlet
nstlist = 20
rlist = 1.2
; Electrostatics / VDW
coulombtype = PME
rcoulomb = 1.2
vdwtype = Cut-off
vdw-modifier = Force-switch
rvdw-switch = 1.0
rvdw = 1.2
; Temperature coupling
tcoupl = V-rescale
tc-grps = SOLU SOLV
tau_t = 0.5 0.5
ref_t = 303.15 303.15
; Parallel/comm
nstcomm = 100
comm_mode = linear
comm_grps = SOLU SOLV
; Velocities
gen-vel = no
; Constraints
constraints = h-bonds
constraint_algorithm = LINCS
lincs_iter = 1
lincs_order = 4
; Pressure coupling
pcoupl = no
; Position restraints
define = -DPOSRE_PROM -DPOSRE_LIG生成并运行 step4.1.3_sidechain_relaxation:
gmx_mpi grompp -c step4.1.2_hydrogen_relaxation.gro -r step4.1.2_hydrogen_relaxation.gro -n index.ndx -f step4.1.3_sidechain_relaxation.mdp -p topol.top -o step4.1.3_sidechain_relaxation.tpr
nohup mpirun -np 16 gmx_mpi mdrun -deffnm step4.1.3_sidechain_relaxation &新建 step4.1.4_protein_relaxation.mdp:
integrator = md
nsteps = 100000 ; 100 ps
dt = 0.001
; Output control
nstxout-compressed = 5000
nstxout = 0
nstvout = 0
nstfout = 0
nstcalcenergy = 100
nstenergy = 500
nstlog = 500
; Neighbor searching
cutoff-scheme = Verlet
nstlist = 20
rlist = 1.2
; Electrostatics / VDW
coulombtype = PME
rcoulomb = 1.2
vdwtype = Cut-off
vdw-modifier = Force-switch
rvdw-switch = 1.0
rvdw = 1.2
; Temperature coupling
tcoupl = V-rescale
tc-grps = SOLU SOLV
tau_t = 0.5 0.5
ref_t = 303.15 303.15
; Parallel/comm
nstcomm = 100
comm_mode = linear
comm_grps = SOLU SOLV
; Velocities
gen-vel = no
; Constraints
constraints = h-bonds
constraint_algorithm = LINCS
lincs_iter = 1
lincs_order = 4
; Pressure coupling
pcoupl = no
; Position restraints
define = -DPOSRE_LIG生成并运行 step4.1.4_protein_relaxation:
gmx_mpi grompp -c step4.1.3_sidechain_relaxation.gro -r step4.1.3_sidechain_relaxation.gro -n index.ndx -f step4.1.4_protein_relaxation.mdp -p topol.top -o step4.1.4_protein_relaxation.tpr
nohup mpirun -np 16 gmx_mpi mdrun -deffnm step4.1.4_protein_relaxation &新建 step4.1.5_system_relaxation.mdp:
integrator = md
nsteps = 100000 ; 100 ps
dt = 0.001
; Output control
nstxout-compressed = 5000
nstxout = 0
nstvout = 0
nstfout = 0
nstcalcenergy = 100
nstenergy = 500
nstlog = 500
; Neighbor searching
cutoff-scheme = Verlet
nstlist = 20
rlist = 1.2
; Electrostatics / VDW
coulombtype = PME
rcoulomb = 1.2
vdwtype = Cut-off
vdw-modifier = Force-switch
rvdw-switch = 1.0
rvdw = 1.2
; Temperature coupling
tcoupl = V-rescale
tc-grps = SOLU SOLV
tau_t = 0.5 0.5
ref_t = 303.15 303.15
; Parallel/comm
nstcomm = 100
comm_mode = linear
comm_grps = SOLU SOLV
; Velocities
gen-vel = no
; Bond constraints
constraints = h-bonds
constraint_algorithm = LINCS
lincs_iter = 1
lincs_order = 4
; Pressure coupling
pcoupl = no
; Position restraints生成并运行 step4.1.5_system_relaxation:
gmx_mpi grompp -c step4.1.4_protein_relaxation.gro -n index.ndx -f step4.1.5_system_relaxation.mdp -p topol.top -o step4.1.5_system_relaxation.tpr
nohup mpirun -np 16 gmx_mpi mdrun -deffnm step4.1.5_system_relaxation &4.2.2 恒压平衡系列
完成短期恒容平衡后,使用 step4.2.1_solvent_relaxation.mdp 进入短期恒压平衡阶段:
integrator = md
nsteps = 100000 ; 100 ps
dt = 0.001
; Output control
nstxout-compressed = 5000
nstxout = 0
nstvout = 0
nstfout = 0
nstcalcenergy = 100
nstenergy = 500
nstlog = 500
; Neighbor searching
cutoff-scheme = Verlet
nstlist = 20
rlist = 1.2
; Electrostatics / VDW
coulombtype = PME
rcoulomb = 1.2
vdwtype = Cut-off
vdw-modifier = Force-switch
rvdw-switch = 1.0
rvdw = 1.2
; Temperature coupling
tcoupl = V-rescale
tc-grps = SOLU SOLV
tau_t = 0.5 0.5
ref_t = 303.15 303.15
; Pressure coupling
pcoupl = C-rescale
pcoupltype = isotropic
tau_p = 5.0
compressibility = 4.5e-5
ref_p = 1.0
; Reference coordinate scaling for restraints
refcoord_scaling = com
; Parallel/comm
nstcomm = 100
comm_mode = linear
comm_grps = SOLU SOLV
; Velocities
gen-vel = no
; Bond constraints
constraints = h-bonds
constraint_algorithm = LINCS
lincs_iter = 1
lincs_order = 4
; Position restraints
define = -DPOSRE_PROA -DPOSRE_LIG生成并运行 step4.2.1_solvent_relaxation:
gmx_mpi grompp -c step4.1.5_system_relaxation.gro -r step4.1.5_system_relaxation.gro -n index.ndx -f step4.2.1_solvent_relaxation.mdp -p topol.top -o step4.2.1_solvent_relaxation.tpr
nohup mpirun -np 16 gmx_mpi mdrun -deffnm step4.2.1_solvent_relaxation &新建 step4.2.2_hydrogen_relaxation.mdp:
integrator = md
nsteps = 100000 ; 100 ps
dt = 0.001
; Output control
nstxout-compressed = 5000
nstxout = 0
nstvout = 0
nstfout = 0
nstcalcenergy = 100
nstenergy = 500
nstlog = 500
; Neighbor searching
cutoff-scheme = Verlet
nstlist = 20
rlist = 1.2
; Electrostatics / VDW
coulombtype = PME
rcoulomb = 1.2
vdwtype = Cut-off
vdw-modifier = Force-switch
rvdw-switch = 1.0
rvdw = 1.2
; Temperature coupling
tcoupl = V-rescale
tc-grps = SOLU SOLV
tau_t = 0.5 0.5
ref_t = 303.15 303.15
; Pressure coupling
pcoupl = C-rescale
pcoupltype = isotropic
tau_p = 5.0
compressibility = 4.5e-5
ref_p = 1.0
; Reference coordinate scaling for restraints
refcoord_scaling = com
; Parallel/comm
nstcomm = 100
comm_mode = linear
comm_grps = SOLU SOLV
; Velocities
gen-vel = no
; Bond constraints
constraints = h-bonds
constraint_algorithm = LINCS
lincs_iter = 1
lincs_order = 4
; Position restraints
define = -DPOSRE_PROH -DPOSRE_LIG生成并运行 step4.2.2_hydrogen_relaxation:
gmx_mpi grompp -c step4.2.1_solvent_relaxation.gro -r step4.2.1_solvent_relaxation.gro -n index.ndx -f step4.2.2_hydrogen_relaxation.mdp -p topol.top -o step4.2.2_hydrogen_relaxation.tpr
nohup mpirun -np 16 gmx_mpi mdrun -deffnm step4.2.2_hydrogen_relaxation &新建 step4.2.3_sidechain_relaxation.mdp:
integrator = md
nsteps = 100000 ; 100 ps
dt = 0.001
; Output control
nstxout-compressed = 5000
nstxout = 0
nstvout = 0
nstfout = 0
nstcalcenergy = 100
nstenergy = 500
nstlog = 500
; Neighbor searching
cutoff-scheme = Verlet
nstlist = 20
rlist = 1.2
; Electrostatics / VDW
coulombtype = PME
rcoulomb = 1.2
vdwtype = Cut-off
vdw-modifier = Force-switch
rvdw-switch = 1.0
rvdw = 1.2
; Temperature coupling
tcoupl = V-rescale
tc-grps = SOLU SOLV
tau_t = 0.5 0.5
ref_t = 303.15 303.15
; Pressure coupling
pcoupl = C-rescale
pcoupltype = isotropic
tau_p = 5.0
compressibility = 4.5e-5
ref_p = 1.0
; Reference coordinate scaling for restraints
refcoord_scaling = com
; Parallel/comm
nstcomm = 100
comm_mode = linear
comm_grps = SOLU SOLV
; Velocities
gen-vel = no
; Bond constraints
constraints = h-bonds
constraint_algorithm = LINCS
lincs_iter = 1
lincs_order = 4
; Position restraints
define = -DPOSRE_PROM -DPOSRE_LIG生成并运行 step4.2.3_sidechain_relaxation:
gmx_mpi grompp -c step4.2.2_hydrogen_relaxation.gro -r step4.2.2_hydrogen_relaxation.gro -n index.ndx -f step4.2.3_sidechain_relaxation.mdp -p topol.top -o step4.2.3_sidechain_relaxation.tpr
nohup mpirun -np 16 gmx_mpi mdrun -deffnm step4.2.3_sidechain_relaxation &新建 step4.2.4_protein_relaxation.mdp:
integrator = md
nsteps = 100000 ; 100 ps
dt = 0.001
; Output control
nstxout-compressed = 5000
nstxout = 0
nstvout = 0
nstfout = 0
nstcalcenergy = 100
nstenergy = 500
nstlog = 500
; Neighbor searching
cutoff-scheme = Verlet
nstlist = 20
rlist = 1.2
; Electrostatics / VDW
coulombtype = PME
rcoulomb = 1.2
vdwtype = Cut-off
vdw-modifier = Force-switch
rvdw-switch = 1.0
rvdw = 1.2
; Temperature coupling
tcoupl = V-rescale
tc-grps = SOLU SOLV
tau_t = 0.5 0.5
ref_t = 303.15 303.15
; Pressure coupling
pcoupl = C-rescale
pcoupltype = isotropic
tau_p = 5.0
compressibility = 4.5e-5
ref_p = 1.0
; Reference coordinate scaling for restraints
refcoord_scaling = com
; Parallel/comm
nstcomm = 100
comm_mode = linear
comm_grps = SOLU SOLV
; Velocities
gen-vel = no
; Bond constraints
constraints = h-bonds
constraint_algorithm = LINCS
lincs_iter = 1
lincs_order = 4
; Position restraints
define = -DPOSRE_LIG生成并运行 step4.2.4_protein_relaxation:
gmx_mpi grompp -c step4.2.3_sidechain_relaxation.gro -r step4.2.3_sidechain_relaxation.gro -n index.ndx -f step4.2.4_protein_relaxation.mdp -p topol.top -o step4.2.4_protein_relaxation.tpr
nohup mpirun -np 16 gmx_mpi mdrun -deffnm step4.2.4_protein_relaxation &新建 step4.2.5_system_relaxation.mdp:
integrator = md
nsteps = 100000 ; 100 ps
dt = 0.001
; Output control
nstxout-compressed = 5000
nstxout = 0
nstvout = 0
nstfout = 0
nstcalcenergy = 100
nstenergy = 500
nstlog = 500
; Neighbor searching
cutoff-scheme = Verlet
nstlist = 20
rlist = 1.2
; Electrostatics / VDW
coulombtype = PME
rcoulomb = 1.2
vdwtype = Cut-off
vdw-modifier = Force-switch
rvdw-switch = 1.0
rvdw = 1.2
; Temperature coupling
tcoupl = V-rescale
tc-grps = SOLU SOLV
tau_t = 0.5 0.5
ref_t = 303.15 303.15
; Pressure coupling
pcoupl = C-rescale
pcoupltype = isotropic
tau_p = 5.0
compressibility = 4.5e-5
ref_p = 1.0
; Reference coordinate scaling for restraints
refcoord_scaling = com
; Parallel/comm
nstcomm = 100
comm_mode = linear
comm_grps = SOLU SOLV
; Velocities
gen-vel = no
; Bond constraints
constraints = h-bonds
constraint_algorithm = LINCS
lincs_iter = 1
lincs_order = 4
; Position restraints生成并运行 step4.2.5_system_relaxation:
gmx_mpi grompp -c step4.2.4_protein_relaxation.gro -n index.ndx -f step4.2.5_system_relaxation.mdp -p topol.top -o step4.2.5_system_relaxation.tpr
nohup mpirun -np 16 gmx_mpi mdrun -deffnm step4.2.5_system_relaxation &4.3 生产模拟阶段
最后基于 step5.1_production.mdp 进行动力学模拟:
integrator = md
nsteps = 5000000 ; 10 ns
dt = 0.002
; Output control
nstxout-compressed = 50000
nstxout = 0
nstvout = 0
nstfout = 0
nstcalcenergy = 100
nstenergy = 1000
nstlog = 1000
; Neighbor searching
cutoff-scheme = Verlet
nstlist = 20
rlist = 1.2
; Electrostatics / VDW
coulombtype = PME
rcoulomb = 1.2
vdwtype = Cut-off
vdw-modifier = Force-switch
rvdw-switch = 1.0
rvdw = 1.2
; Temperature coupling
tcoupl = V-rescale
tc_grps = SOLU SOLV
tau_t = 0.5 0.5
ref_t = 303.15 303.15
; Pressure coupling
pcoupl = Parrinello-Rahman
pcoupltype = isotropic
tau_p = 5.0
compressibility = 4.5e-5
ref_p = 1.0
; Parallel/comm
nstcomm = 100
comm_mode = linear
comm_grps = SOLU SOLV
; Velocities
gen-vel = no
; Bond constraints
constraints = h-bonds
constraint_algorithm = LINCS
lincs_iter = 2
lincs_order = 6生成并运行 step5.1_production:
gmx_mpi grompp -c step4.2.5_system_relaxation.gro -n index.ndx -f step5.1_production.mdp -p topol.top -o step5.1_production.tpr
nohup mpirun -np 16 gmx_mpi mdrun -deffnm step5.1_production &至此,分子动力学正常运行,待分子动力学运行结束后分析轨迹文件即可,相关分析过程留待后续介绍。
发表回复