
Gromacs 是一款广泛用于药物设计领域的开源分子动力学模拟软件。在前期研究中,它通常部署于高性能计算集群。然而,鉴于苹果公司推出的 Apple Silicon 芯片展现出的卓越性能,这里以 2022 年发布的 Mac Studio 为例,详细阐述 Gromacs 在 Apple Silicon 平台上的部署与运行策略。
一、安装考量
1.1 GPU 加速问题
由于 macOS 已逐步淘汰 OpenCL,转而采用 Metal 作为 GPU 支持方案,但当前版本的 Gromacs 尚未实现对 Metal 的支持。尽管可以通过 MoltenVK(Vulkan 到 Metal 的转换层)在 macOS 上运行 Vulkan,但 Gromacs 对 macOS Vulkan 后端的支持、稳定性及性能并非官方主流推荐。因此,为确保系统的稳定性并充分发挥 CPU 的最高性能,我们选择在原生环境下禁用 GPU 加速(-DGMX_GPU=OFF)。
1.2 MPI 方案的选择
鉴于我们的部署环境并非多节点集群,为了充分利用 M1 Ultra 的 20 个 CPU 核心,同时简化安装流程,我们决定采用系统自带的 MPI(Apple MPICH 的裁剪版本),而无需额外安装 OpenMPI。
二、工具安装
2.1 安装必要的基础工具
使用 Homebrew 安装编译 Gromacs 所需的基础工具:
brew install cmake fftw gcc2.2 设置编译环境
配置编译所需的环境变量,并重新加载配置文件:
export CC=/opt/homebrew/bin/gcc-14
export CXX=/opt/homebrew/bin/g++-14
export PATH="/opt/homebrew/bin:$PATH"
source ~/.zshrc三、Gromacs 安装
3.1 下载 Gromacs 安装包
前往 gromacs.org ,获取最新版的 Gromacs 安装包的下载链接:
下载并解压安装包:
wget https://ftp.gromacs.org/gromacs/gromacs-2025.3.tar.gz
tar -zxvf gromacs-2025.3.tar
cd gromacs-2025.3
mkdir build && cd build3.2 编译配置
执行以下命令进行编译配置:
cmake .. -DCMAKE_BUILD_TYPE=Release -DGMX_FFT_LIBRARY=fftw3 -DGMX_MPI=ON -DGMX_OPENMP=OFF -DGMX_SIMD=ARM_NEON_ASIMD -DGMX_DOUBLE=OFF -DGMX_GPU=OFF -DGMX_USE_RDTSCP=OFF -DCMAKE_INSTALL_PREFIX=$HOME/gromacs -DCMAKE_C_COMPILER=clang -DCMAKE_CXX_COMPILER=clang++ -DGMX_BUILD_OWN_FFTW=ON说明
-DGMX_SIMD=ARM_NEON_ASIMD:在 ARM64 架构上,为获得最佳短距核(non-bonded)性能,推荐显式设置此选项。-DGMX_MPI=ON:由于 Apple 自带的 Clang 编译器显式禁用了 OpenMP,启用 MPI 并行化可替代 OpenMP 的功能。在 M1 Ultra 的 16-20 核环境下,通过 MPI ranks 分布到 P-cores 是系统推荐的稳定方案。-DGMX_USE_RDTSCP=OFF:RDTSCP 是性能测试和精确计时的一种优化手段,在 Intel/AMD x86 CPU + Linux 环境下表现良好,但 ARM 架构不支持 RDTSCP 指令。GROMACS 会回退到std::chrono或 macOS 提供的mach_absolute_time()。在 M1 Ultra 上,这些 API 的性能已经非常接近 RDTSCP,因此选择禁用。
3.3 编译与检查
完成编译配置后,执行以下命令进行编译和检查:
make -j$(sysctl -n hw.ncpu) && make check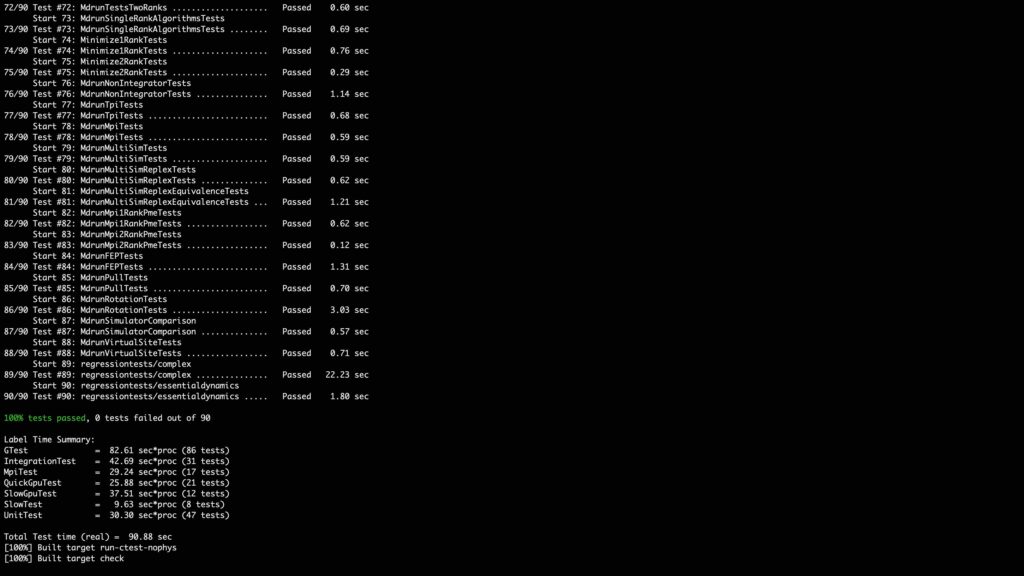
3.4 安装 Gromacs
编译检查通过后,执行安装命令:
make install
3.5 设置环境变量
安装完成后,设置环境变量并重新索引:
echo 'export PATH=$HOME/gromacs/bin:$PATH' >> ~/.zshrc
source ~/.zshrc四、验证安装
4.1 Gromacs 安装情况检查
执行以下命令验证 Gromacs 是否安装成功:
(base) hujunchi@build % gmx_mpi mdrun -h
:-) GROMACS - gmx mdrun, 2025.3 (-:
Executable: /Users/hujunchi/gromacs/bin/gmx_mpi
Data prefix: /Users/hujunchi/gromacs
Working dir: /Users/hujunchi/Downloads/water-cut1.0_bare_hbonds/0003
Command line:
gmx_mpi mdrun -h
SYNOPSIS
gmx mdrun [-s [<.tpr>]] [-cpi [<.cpt>]] [-table [<.xvg>]] [-tablep [<.xvg>]]
[-tableb [<.xvg> [...]]] [-rerun [<.xtc/.trr/...>]] [-ei [<.edi>]]
[-multidir [<dir> [...]]] [-awh [<.xvg>]] [-plumed [<.dat>]]
[-membed [<.dat>]] [-mp [<.top>]] [-mn [<.ndx>]]
[-o [<.trr/.cpt/...>]] [-x [<.xtc/.tng>]] [-cpo [<.cpt>]]
[-c [<.gro/.g96/...>]] [-e [<.edr>]] [-g [<.log>]] [-dhdl [<.xvg>]]
[-field [<.xvg>]] [-tpi [<.xvg>]] [-tpid [<.xvg>]] [-eo [<.xvg>]]
[-px [<.xvg>]] [-pf [<.xvg>]] [-ro [<.xvg>]] [-ra [<.log>]]
[-rs [<.log>]] [-rt [<.log>]] [-mtx [<.mtx>]] [-if [<.xvg>]]
[-swap [<.xvg>]] [-deffnm <string>] [-xvg <enum>] [-dd <vector>]
[-ddorder <enum>] [-npme <int>] [-nt <int>] [-ntmpi <int>]
[-ntomp <int>] [-ntomp_pme <int>] [-pin <enum>] [-pinoffset <int>]
[-pinstride <int>] [-gpu_id <string>] [-gputasks <string>]
[-[no]ddcheck] [-rdd <real>] [-rcon <real>] [-dlb <enum>]
[-dds <real>] [-nb <enum>] [-nstlist <int>] [-[no]tunepme]
[-pme <enum>] [-pmefft <enum>] [-bonded <enum>] [-update <enum>]
[-[no]v] [-pforce <real>] [-[no]reprod] [-cpt <real>] [-[no]cpnum]
[-[no]append] [-nsteps <int>] [-maxh <real>] [-replex <int>]
[-nex <int>] [-reseed <int>]
DESCRIPTION
gmx mdrun is the main computational chemistry engine within GROMACS.
Obviously, it performs Molecular Dynamics simulations, but it can also perform
Stochastic Dynamics, Energy Minimization, test particle insertion or
(re)calculation of energies. Normal mode analysis is another option. In this
case mdrun builds a Hessian matrix from single conformation. For usual Normal
Modes-like calculations, make sure that the structure provided is properly
energy-minimized. The generated matrix can be diagonalized by gmx nmeig.4.2 实际运行测试
访问 gromacs.org 获取测试用 Gromacs 文件,并下载:
cd ..
mkdir test && cd test
wget https://ftp.gromacs.org/pub/benchmarks/water_bare_hbonds.tar.gz
gzip -zxvf water_bare_hbonds.tar.gz
cd water-cut1.0_bare_hbonds/0003使用 .gro 和 .top 文件生成 .tpr 文件:
gmx_mpi grompp -f pme.mdp -c conf.gro -p topol.top -o topol.tpr考虑到 M1 Ultra 的异构架构(包含 16 个大核 P-core 和 4 个小核 E-core),为避免负载不平衡,我们选择仅使用 16 个大核进行并行计算,并由系统自行分配核心:
(base) hujunchi@build % mpirun -np 16 gmx_mpi mdrun -s topol.tpr -deffnm test
:-) GROMACS - gmx mdrun, 2025.3 (-:
Executable: /Users/hujunchi/gromacs/bin/gmx_mpi
Data prefix: /Users/hujunchi/gromacs
Working dir: /Users/hujunchi/Downloads/water-cut1.0_bare_hbonds/0003
Command line:
gmx_mpi mdrun -s topol.tpr -deffnm test
Reading file topol.tpr, VERSION 2025.3 (single precision)
Changing nstlist from 10 to 40, rlist from 1 to 1.099
Using 16 MPI processes
starting mdrun 'Water'
33000 steps, 66.0 ps.
Writing final coordinates.
Dynamic load balancing report:
DLB got disabled because it was unsuitable to use.
Average load imbalance: 15.5%.
The balanceable part of the MD step is 52%, load imbalance is computed from this.
Part of the total run time spent waiting due to load imbalance: 8.1%.
NOTE: 8.1 % of the available CPU time was lost due to load imbalance
in the domain decomposition.
You can consider manually changing the decomposition (option -dd);
e.g. by using fewer domains along the box dimension in which there is
considerable inhomogeneity in the simulated system.
Core t (s) Wall t (s) (%)
Time: 117.582 7.349 1600.0
(ns/day) (hour/ns)
Performance: 775.974 0.031
GROMACS reminds you: "There is no such thing as free energy. Anyone who advocates it does not know what he is talking about." (Alireza Haghighat)注意
由于 M1 Ultra 的异构架构特性,OpenMPI 可能会将部分进程绑定到 E-core,从而导致性能下降。为避免这种情况,可以使用 --bind-to none 选项显式关闭自动绑核,使系统调度器倾向于将负载分配到 P-core 上:
mpirun -np 16 --bind-to none gmx_mpi mdrun -s topol.tpr -deffnm test至此,Gromacs 在 M1 Ultra 上的安装与调试工作全部完成。
发表回复